publications
publications by categories in reversed chronological order.
2025
- Nat. Commun.
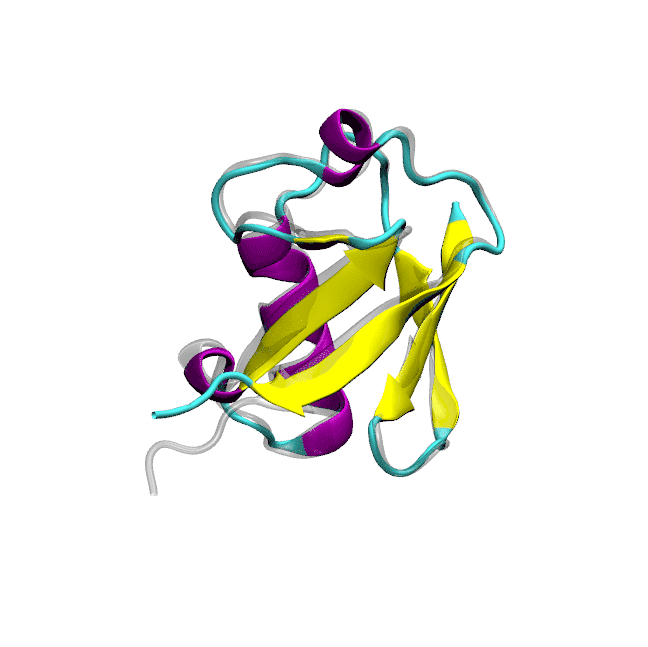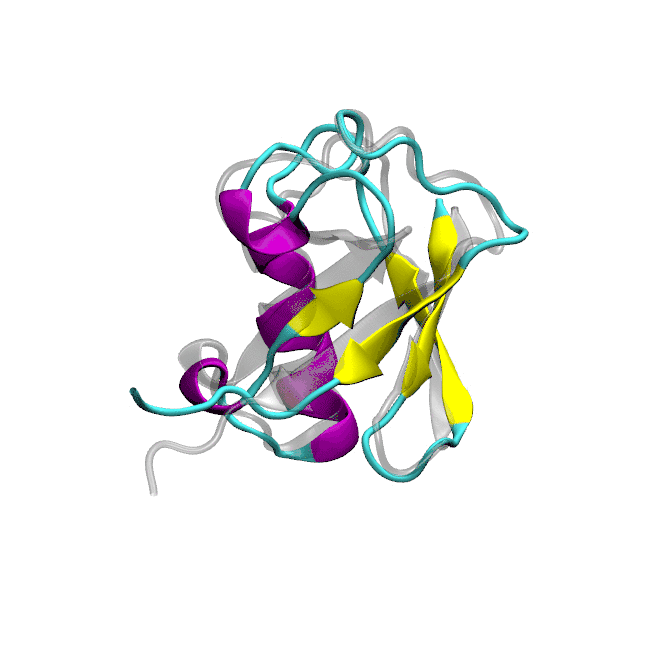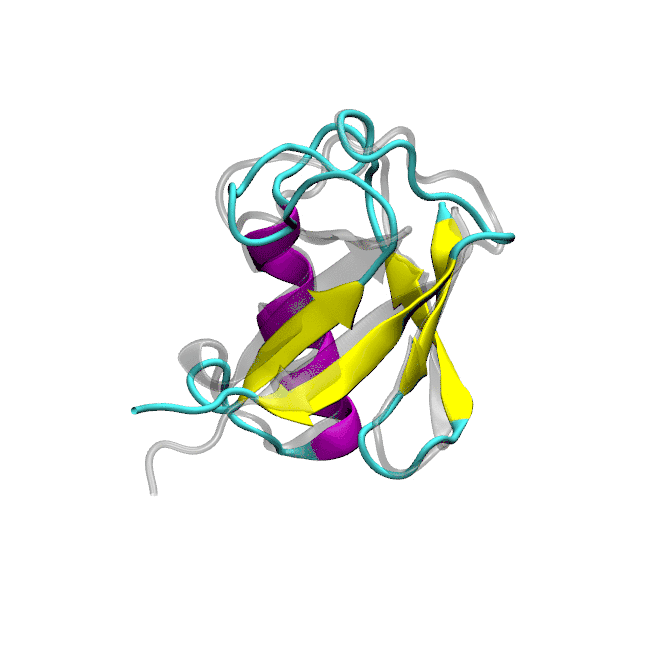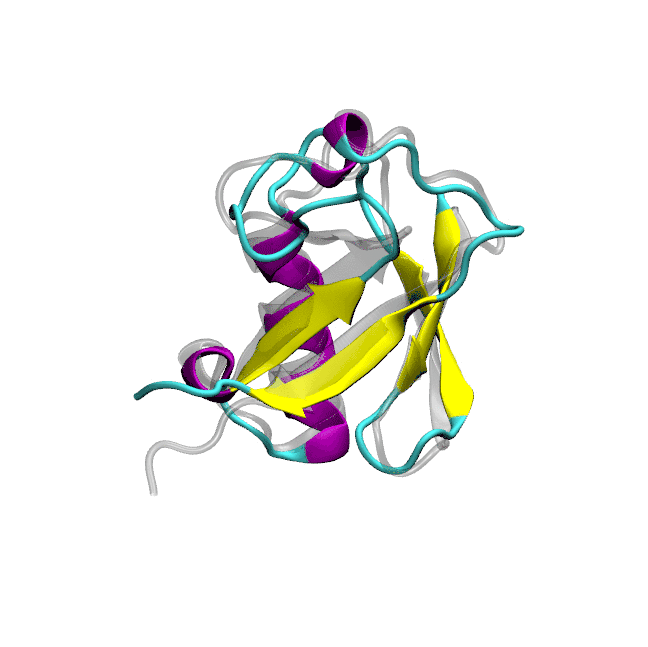 Optimized protein-water interactions and torsional refinements yield balanced atomistic protein force fieldsTien Minh Phan, Priyesh Mohanty, and Jeetain MittalNature Communications, 2025
Optimized protein-water interactions and torsional refinements yield balanced atomistic protein force fieldsTien Minh Phan, Priyesh Mohanty, and Jeetain MittalNature Communications, 2025All-atom molecular dynamics (MD) simulations based on physics-based force fields serve as an essential complement to experiments for investigating protein structure, dynamics, and interactions. Despite significant advances in force field parameterization, achieving a consistent balance of molecular interactions that stabilize folded proteins while accurately capturing the conformational dynamics of intrinsically disordered polypeptides in solution remains challenging. In this work, we introduce two refined force fields which incorporate either a selective upscaling of protein-water interactions or targeted improvements to backbone torsional sampling: (i) amber ff03w-sc, and (ii) amber ff99SBws-STQ′. Extensive validation against small-angle X-ray scattering (SAXS) and nuclear magnetic resonance (NMR) spectroscopy observables revealed that both force fields accurately reproduced the chain dimensions and secondary structure propensities of IDPs. Importantly, both force fields also maintained the stability of single-chain folded proteins and protein-protein complexes over microsecond-timescale simulations. Overall, our refinement strategies result in transferable force fields with improved accuracy for simulating diverse protein systems, ranging from folded domains to IDPs and protein-protein complexes.
@article{phan2025optimized, title = {Optimized protein-water interactions and torsional refinements yield balanced atomistic protein force fields}, author = {Phan, Tien Minh and Mohanty, Priyesh and Mittal, Jeetain}, journal = {Nature Communications}, volume = {16}, number = {1}, pages = {10562}, year = {2025}, publisher = {Nature Publishing Group UK London}, dimensions = {1195414776}, doi = {https://doi.org/10.1038/s41467-025-65603-4}, url = {https://doi.org/10.1038/s41467-025-65603-4}, } - JPCB
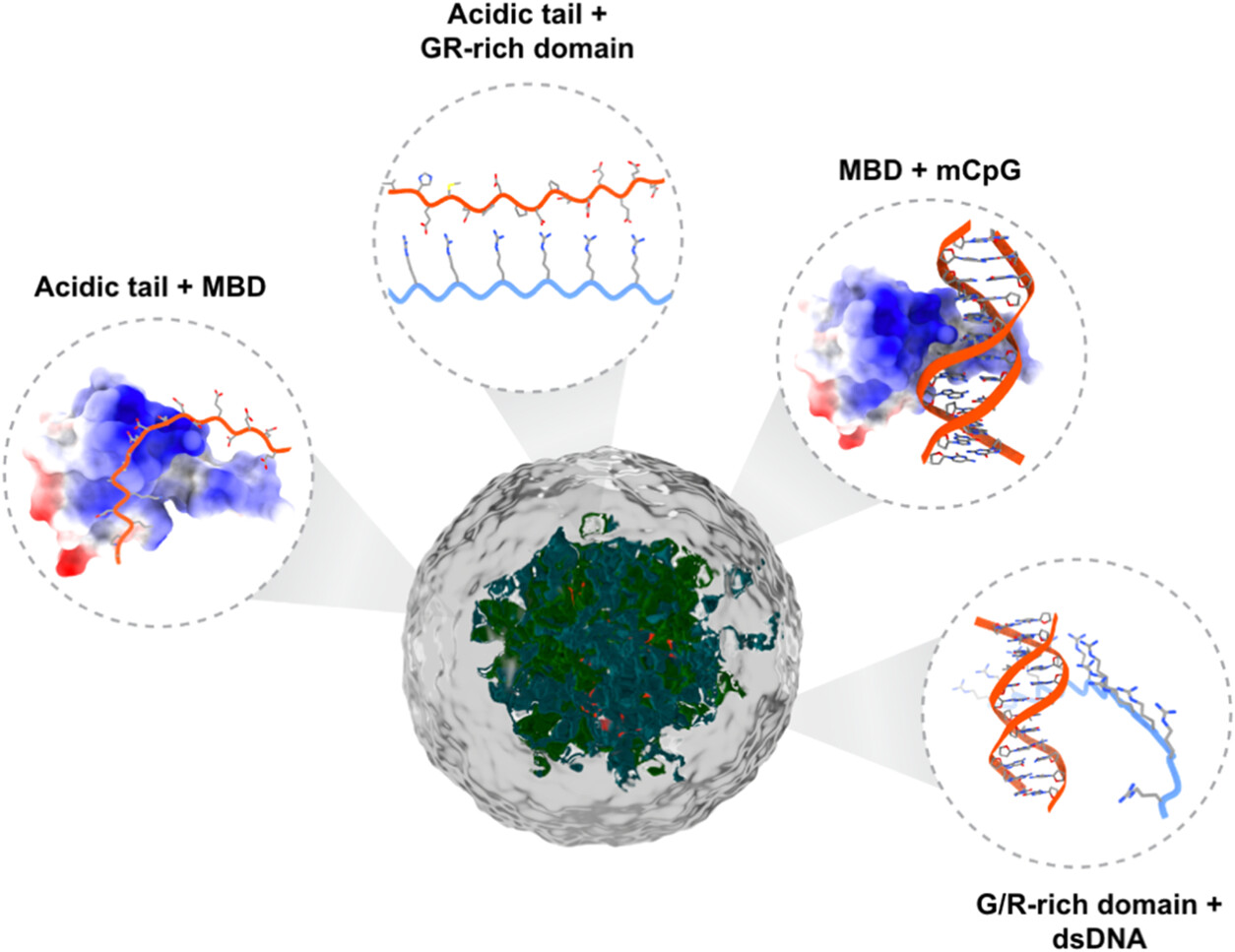 Uncovering the molecular interactions underlying MBD2 and MBD3 phase separationNicole Maurici, Tien M Phan, Jessica L Henty-Ridilla, and 3 more authorsThe Journal of Physical Chemistry B, 2025
Uncovering the molecular interactions underlying MBD2 and MBD3 phase separationNicole Maurici, Tien M Phan, Jessica L Henty-Ridilla, and 3 more authorsThe Journal of Physical Chemistry B, 2025Chromatin organization controls DNA’s accessibility to regulatory factors to influence gene expression. Heterochromatin, or transcriptionally silent chromatin enriched in methylated DNA and methylated histone tails, self-assembles through multivalent interactions with its associated proteins into a condensed, but dynamic state. Liquid–liquid phase separation (LLPS) of key heterochromatin regulators, such as heterochromatin protein 1 (HP1), plays an essential role in heterochromatin assembly and function. Methyl-CpG-binding protein 2 (MeCP2), the most studied member of the methyl-CpG-binding domain (MBD) family of proteins, has been recently shown to undergo LLPS in the absence and presence of methylated DNA. These studies provide a new mechanistic framework for understanding the role of methylated DNA and its readers in heterochromatin formation. However, the details of the molecular interactions by which other MBD family members undergo LLPS to mediate genome organization and transcriptional regulation are not fully understood. Here, we focus on two MBD proteins, MBD2 and MBD3, that have distinct but interdependent roles in gene regulation. Using an integrated computational and experimental approach, we uncover the homotypic and heterotypic interactions governing MBD2 and MBD3 phase separation and DNA’s influence on this process. We show that despite sharing the highest sequence identity and structural homology among all the MBD protein family members, MBD2 and MBD3 exhibit differing residue patterns resulting in distinct phase separation mechanisms. Understanding the molecular underpinnings of MBD protein condensation offers insights into the higher-order, LLPS-mediated organization of heterochromatin.
@article{maurici2025uncovering, title = {Uncovering the molecular interactions underlying MBD2 and MBD3 phase separation}, author = {Maurici, Nicole and Phan, Tien M and Henty-Ridilla, Jessica L and Kim, Young C and Mittal, Jeetain and Bah, Alaji}, journal = {The Journal of Physical Chemistry B}, volume = {129}, number = {23}, pages = {5728--5743}, year = {2025}, doi = {https://doi.org/10.1021/acs.jpcb.5c02741}, url = {https://doi.org/10.1021/acs.jpcb.5c02741}, cover = {https://pubs.acs.org/toc/jpcbfk/129/23}, dimensions = {1188588975}, publisher = {ACS Publications} } - JACS
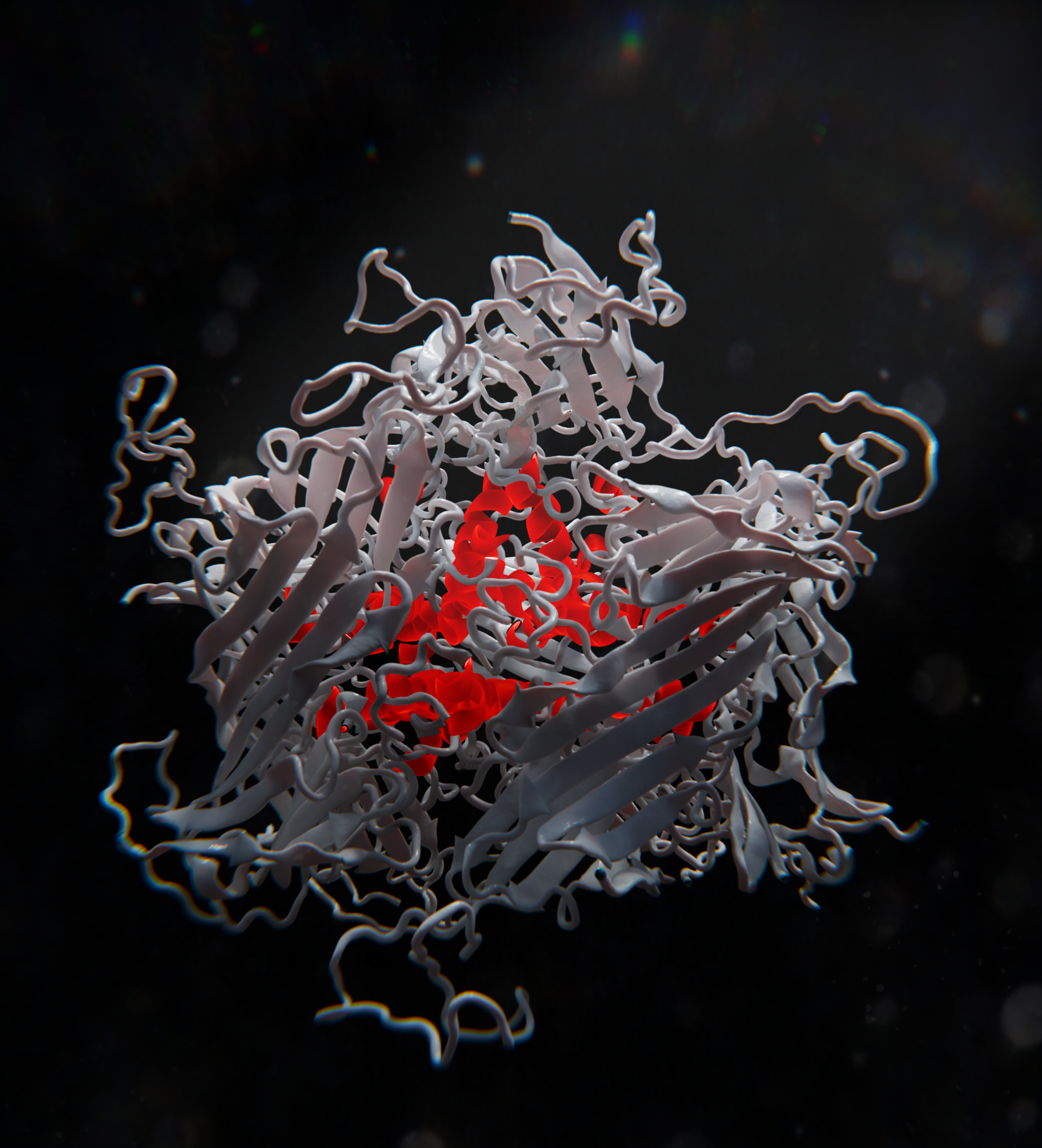 Capturing the Conformational Heterogeneity of HSPB1 Chaperone Oligomers at Atomic ResolutionRaymond F Berkeley, Alexander P Plonski, Tien M Phan, and 6 more authorsJournal of the American Chemical Society, 2025
Capturing the Conformational Heterogeneity of HSPB1 Chaperone Oligomers at Atomic ResolutionRaymond F Berkeley, Alexander P Plonski, Tien M Phan, and 6 more authorsJournal of the American Chemical Society, 2025Small heat shock proteins (sHSPs), including HSPB1, are essential regulators of cellular proteostasis that interact with unfolded and partially folded proteins to prevent aberrant misfolding and aggregation. These proteins fulfill a similar role in biological condensates, where they interact with intrinsically disordered proteins to modulate their liquid–liquid and liquid-to-solid phase transitions. Characterizing the sHSP structure, dynamics, and client interactions is challenging due to their partially disordered nature, their tendency to form polydisperse oligomers, and their diverse range of clients. In this work, we leverage various biophysical methods, including fast 1H-based magic angle spinning (MAS) NMR spectroscopy, molecular dynamics (MD) simulations, and modeling, to shed new light on the structure and dynamics of HSPB1 oligomers. Using split-intein-mediated segmental labeling, we provide unambiguous evidence that in the oligomer context, the N-terminal domain (NTD) of HSPB1 is rigid and adopts an ensemble of heterogeneous conformations, the α-Crystallin domain (ACD) forms dimers and experiences multiple distinct local environments, while the C-terminal domain (CTD) remains highly dynamic. Our computational models suggest that the NTDs participate in extensive NTD–NTD and NTD–ACD interactions and are sequestered within the oligomer interior. We further demonstrate that HSPB1 higher order oligomers disassemble into smaller oligomeric species in the presence of a client protein and that an accessible NTD is essential for HSPB1 partitioning into condensates and interactions with client proteins. Our integrated approach provides a high-resolution view of the complex oligomeric landscape of HSPB1 and sheds light on the elusive network of interactions that underlies the function of HSPB1 in biological condensates.
@article{berkeley2025capturing, title = {Capturing the Conformational Heterogeneity of HSPB1 Chaperone Oligomers at Atomic Resolution}, author = {Berkeley, Raymond F and Plonski, Alexander P and Phan, Tien M and Grohe, Kristof and Becker, Lukas and Wegner, Sebastian and Herzik Jr, Mark A and Mittal, Jeetain and Debelouchina, Galia T}, journal = {Journal of the American Chemical Society}, volume = {147}, number = {18}, pages = {15181--15194}, year = {2025}, publisher = {ACS Publications}, doi = {https://doi.org/10.1021/jacs.4c18668}, url = {https://doi.org/10.1021/jacs.4c18668}, cover = {https://pubs.acs.org/toc/jacsat/147/18}, dimensions = {1187020265}, } - Mol. Cell
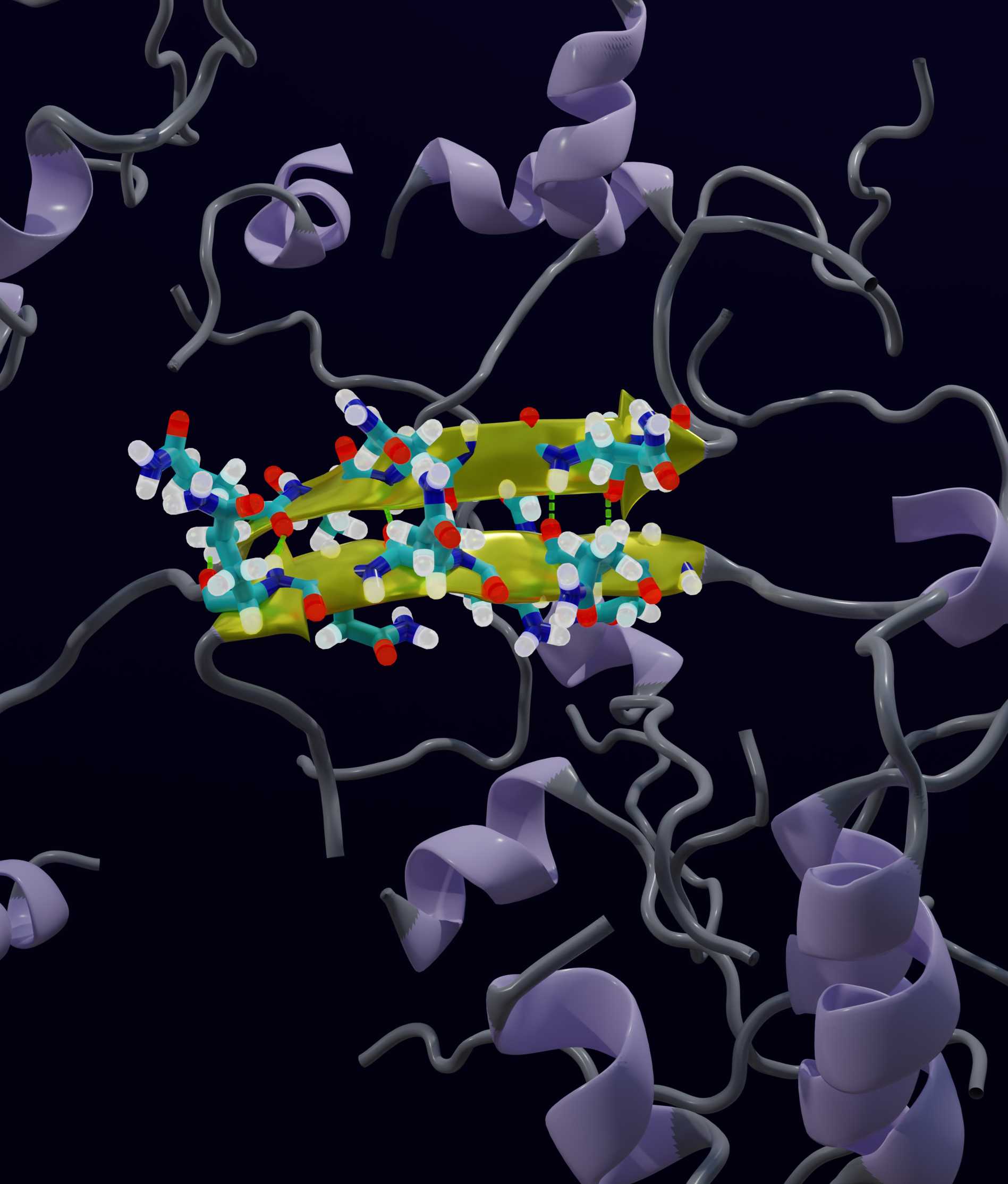
 A disordered linker in the Polycomb protein Polyhomeotic tunes phase separation and oligomerizationTim M Gemeinhardt, Roshan M Regy, Tien M Phan, and 8 more authorsMolecular Cell, 2025
A disordered linker in the Polycomb protein Polyhomeotic tunes phase separation and oligomerizationTim M Gemeinhardt, Roshan M Regy, Tien M Phan, and 8 more authorsMolecular Cell, 2025Biomolecular condensates are increasingly recognized as key regulators of chromatin organization, yet how their formation and properties arise from protein sequences remains incompletely understood. Cross-species comparisons can reveal both conserved functions and significant evolutionary differences. Here, we integrate in vitro reconstitution, molecular dynamics simulations, and cell-based assays to examine how Drosophila and human variants of Polyhomeotic (Ph)—a subunit of the PRC1 chromatin regulatory complex—drive condensate formation through their sterile alpha motif (SAM) oligomerization domains. We identify divergent interactions between SAM and the disordered linker connecting it to the rest of Ph. These interactions enhance oligomerization and modulate both the formation and properties of reconstituted condensates. Oligomerization influences condensate dynamics but minimally impacts condensate formation. Linker-SAM interactions also affect condensate formation in Drosophila and human cells and growth in Drosophila imaginal discs. Our findings show how evolutionary changes in disordered linkers can fine-tune condensate properties, providing insights into sequence-function relationships.
@article{gemeinhardt2025disordered, title = {A disordered linker in the Polycomb protein Polyhomeotic tunes phase separation and oligomerization}, author = {Gemeinhardt, Tim M and Regy, Roshan M and Phan, Tien M and Pal, Nanu and Sharma, Jyoti and Senkovich, Olga and Mendiola, Andrea J and Ledterman, Heather J and Henrickson, Amy and Lopes, Daniel and others}, journal = {Molecular Cell}, volume = {85}, number = {11}, pages = {2128--2146}, year = {2025}, publisher = {Elsevier}, doi = {https://doi.org/10.1016/j.molcel.2025.05.008}, url = {https://doi.org/10.1016/j.molcel.2025.05.008}, dimensions = {1189183571}, } - Adv. Sci.
 Transient Interdomain Interactions Modulate the Monomeric Structural Ensemble and Self-Assembly of Huntingtin Exon 1Priyesh Mohanty, Tien Minh Phan, and Jeetain MittalAdvanced Science, 2025
Transient Interdomain Interactions Modulate the Monomeric Structural Ensemble and Self-Assembly of Huntingtin Exon 1Priyesh Mohanty, Tien Minh Phan, and Jeetain MittalAdvanced Science, 2025Polyglutamine (polyQ) tract length expansion (≥ 36 residues) within the N-terminal exon-1 of Huntingtin (Httex1) leads to Huntington’s disease, a neurodegenerative condition marked by the presence of intranuclear Htt inclusions. Notably, the polyQ tract in Httex1 is flanked by an N-terminal coiled-coil domain -N17 (17 amino acids), which promotes the formation of soluble oligomers and brings the aggregation-prone polyQ tracts in close proximity. However, the molecular mechanisms underlying the conversion of soluble oligomers into insoluble β-rich aggregates with increasing polyQ length, remain unclear. In this study, extensive atomistic molecular dynamics (MD) simulations (aggregate time ≈0.7 milliseconds) are performed to uncover the interplay between structural transformation and domain "cross-talk" on the conformational ensemble and oligomerization of Httex1 due to polyQ expansion. Notably, MD-derived ensembles of N17-Qn-P5 monomers validated against NMR indicated that in addition to elevated α-helicity, polyQ expansion also favored transient, interdomain (N17/polyQ) interactions which resulted in the emergence of β-sheet conformations. Further, interdomain interactions modulated the stability of N17-mediated polyQ dimers and promoted a heterogeneous dimerization landscape. Finally, it is observed that the intact C-terminal proline-rich domain (PRD) promoted condensation of Httex1 through self-interactions involving its P10/P11 tracts while also interacting with N17 to suppress its α-helicity.
@article{mohanty2025transient, title = {Transient Interdomain Interactions Modulate the Monomeric Structural Ensemble and Self-Assembly of Huntingtin Exon 1}, author = {Mohanty, Priyesh and Phan, Tien Minh and Mittal, Jeetain}, journal = {Advanced Science}, volume = {12}, number = {27}, pages = {2501462}, year = {2025}, publisher = {Wiley Online Library}, dimensions = {1188099306}, doi = {https://doi.org/10.1002/advs.202501462}, url = {https://doi.org/10.1002/advs.202501462}, } - JPCB
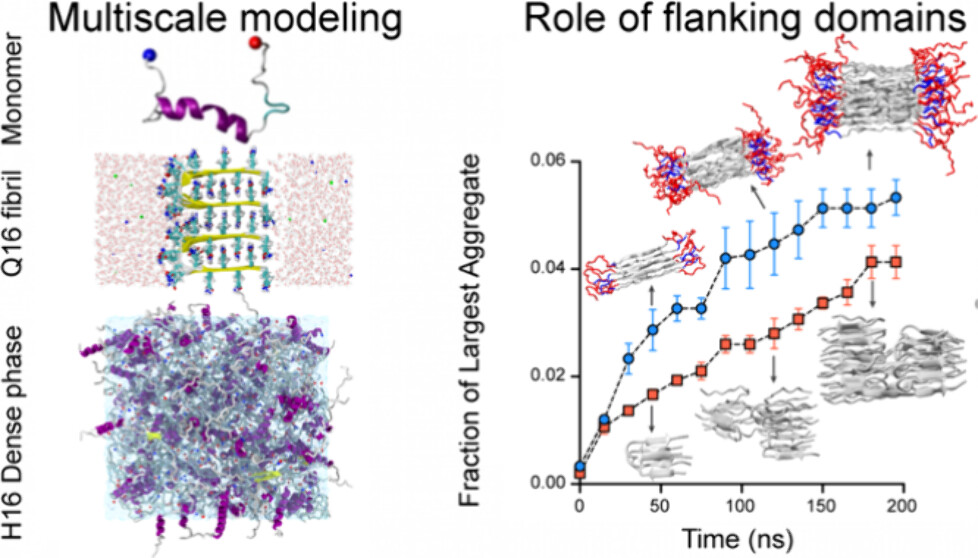 Multiscale simulations elucidate the mechanism of polyglutamine aggregation and the role of flanking domains in fibril polymorphismAvijeet Kulshrestha, Tien Minh Phan, Azamat Rizuan, and 2 more authorsThe Journal of Physical Chemistry B, 2025
Multiscale simulations elucidate the mechanism of polyglutamine aggregation and the role of flanking domains in fibril polymorphismAvijeet Kulshrestha, Tien Minh Phan, Azamat Rizuan, and 2 more authorsThe Journal of Physical Chemistry B, 2025Protein aggregation, which is implicated in aging and neurodegenerative diseases, typically involves a transition from soluble monomers and oligomers to insoluble fibrils. Polyglutamine (polyQ) tracts in proteins can form amyloid fibrils, which are linked to polyQ diseases, including Huntington’s disease (HD), where the length of the polyQ tract inversely correlates with the age of onset. Despite significant research on the mechanisms of Httex1 aggregation, atomistic information regarding the intermediate stages of its fibrillation and the morphological characteristics of the end-state amyloid fibrils remains limited. Recently, molecular dynamics (MD) simulations based on a hybrid multistate structure-based model, Multi-eGO, have shown promise in capturing the kinetics and mechanism of amyloid fibrillation with high computational efficiency while achieving qualitative agreement with experiments. Here, we utilize the multi-eGO simulation methodology to study the mechanism and kinetics of polyQ fibrillation and the effect of the N17 flanking domain of Huntingtin protein. Aggregation simulations of polyQ produced highly heterogeneous amyloid fibrils with variable-width branched morphologies by incorporating combinations of β-turn, β-arc, and β-strand structures, while the presence of the N17 flanking domain reduces amyloid fibril heterogeneity by favoring β-strand conformations. Our simulations reveal that the presence of N17 domain enhanced aggregation kinetics by promoting the formation of large, structurally stable oligomers. Furthermore, the early-stage aggregation process involves two distinct mechanisms: backbone interactions driving β-sheet formation and side-chain interdigitation. Overall, our study provides detailed insights into fibrillation kinetics, mechanisms, and end-state polymorphism associated with Httex1 amyloid aggregation. SIGNIFICANCE STATEMENT: Polyglutamine (polyQ) aggregation is central to Huntington’s disease and related neurodegenerative disorders. Despite extensive experimental efforts, a complete molecular understanding of this process-from early aggregation events to the origins of fibril polymorphism-has remained elusive, with varied interpretations of complex fibril architectures. Through multiscale simulations, we reveal how polyQ fibrils adopt diverse tertiary and quaternary structures and demonstrate how the N-terminal flanking domain (N17) modulates fibril architecture and accelerates aggregation. Our hybrid multi-eGO simulations capture early-stage fibrillation kinetics and identify distinct structural polymorphs that align with experimental observations. This work provides a molecular framework for understanding amyloid polymorphism and illuminates the role of flanking domains in shaping aggregation pathways-offering valuable insights for therapeutic strategies targeting early toxic intermediates.
@article{kulshrestha2025multiscale, title = {Multiscale simulations elucidate the mechanism of polyglutamine aggregation and the role of flanking domains in fibril polymorphism}, author = {Kulshrestha, Avijeet and Phan, Tien Minh and Rizuan, Azamat and Mohanty, Priyesh and Mittal, Jeetain}, journal = {The Journal of Physical Chemistry B}, number = {ASAP}, pages = {ASAP}, year = {2025}, publisher = {ACS Publications}, dimensions = {1189001894}, doi = {https://doi.org/10.1021/acs.jpcb.5c06627}, url = {https://doi.org/10.1021/acs.jpcb.5c06627}, }
2024
- eLife
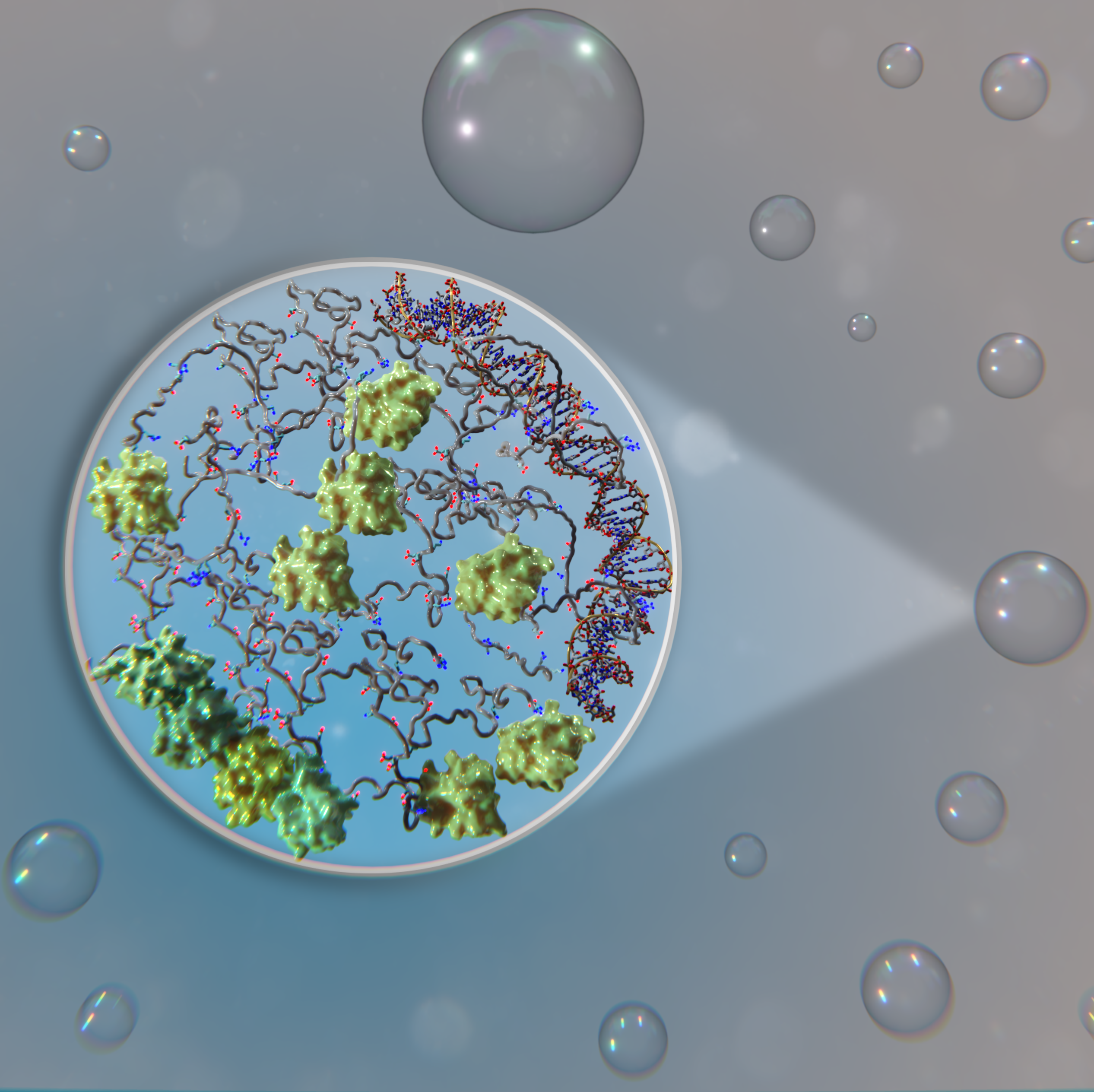
 Interplay between charge distribution and DNA in shaping HP1 paralog phase separation and localizationTien M Phan, Young C Kim, Galia T Debelouchina, and 1 more authorElife, 2024
Interplay between charge distribution and DNA in shaping HP1 paralog phase separation and localizationTien M Phan, Young C Kim, Galia T Debelouchina, and 1 more authorElife, 2024The heterochromatin protein 1 (HP1) family is a crucial component of heterochromatin with diverse functions in gene regulation, cell cycle control, and cell differentiation. In humans, there are three paralogs, HP1α, HP1β, and HP1γ, which exhibit remarkable similarities in their domain architecture and sequence properties. Nevertheless, these paralogs display distinct behaviors in liquid-liquid phase separation (LLPS), a process linked to heterochromatin formation. Here, we employ a coarse-grained simulation framework to uncover the sequence features responsible for the observed differences in LLPS. We highlight the significance of the net charge and charge patterning along the sequence in governing paralog LLPS propensities. We also show that both highly conserved folded and less-conserved disordered domains contribute to the observed differences. Furthermore, we explore the potential co-localization of different HP1 paralogs in multicomponent assemblies and the impact of DNA on this process. Importantly, our study reveals that DNA can significantly reshape the stability of a minimal condensate formed by HP1 paralogs due to competitive interactions of HP1α with HP1β and HP1γ versus DNA. In conclusion, our work highlights the physicochemical nature of interactions that govern the distinct phase-separation behaviors of HP1 paralogs and provides a molecular framework for understanding their role in chromatin organization.
@article{phan2024interplay, title = {Interplay between charge distribution and DNA in shaping HP1 paralog phase separation and localization}, author = {Phan, Tien M and Kim, Young C and Debelouchina, Galia T and Mittal, Jeetain}, journal = {Elife}, volume = {12}, pages = {RP90820}, year = {2024}, publisher = {eLife Sciences Publications Limited}, doi = {https://doi.org/10.7554/eLife.90820.3}, url = {https://doi.org/10.7554/eLife.90820.3}, dimensions = {1164747815} } - JPCB
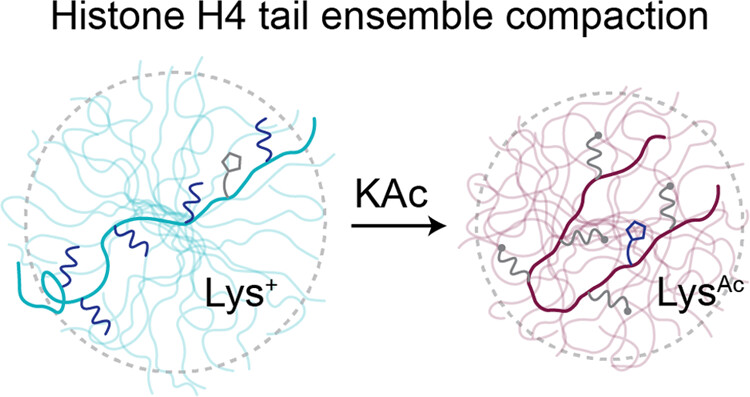 Acetylation-Dependent Compaction of the Histone H4 Tail EnsembleSophia M Dewing, Tien M Phan, Emma J Kraft, and 2 more authorsThe Journal of Physical Chemistry B, 2024
Acetylation-Dependent Compaction of the Histone H4 Tail EnsembleSophia M Dewing, Tien M Phan, Emma J Kraft, and 2 more authorsThe Journal of Physical Chemistry B, 2024Acetylation of the histone H4 tail (H4Kac) has been established as a significant regulator of chromatin architecture and accessibility; however, the molecular mechanisms that underlie these observations remain elusive. Here, we characterize the ensemble features of the histone H4 tail and determine how they change following acetylation on specific sets of lysine residues. Our comprehensive account is enabled by a robust combination of experimental and computational biophysical methods that converge on molecular details including conformer size, intramolecular contacts, and secondary structure propensity. We find that acetylation significantly alters the chemical environment of basic patch residues (16-20) and leads to tail compaction that is partially mediated by transient intramolecular contacts established between the basic patch and N-terminal amino acids. Beyond acetylation, we identify that the protonation state of H18, which is affected by the acetylation state, is a critical regulator of ensemble characteristics, highlighting the potential for interplay between the sequence context and post-translational modifications to define the ensemble features of intrinsically disordered regions. This study elucidates molecular details that could link H4Kac with the regulation of chromatin architecture, illuminating a small piece of the complex network of molecular mechanisms underlying the histone code hypothesis.
@article{dewing2024acetylation, title = {Acetylation-Dependent Compaction of the Histone H4 Tail Ensemble}, author = {Dewing, Sophia M and Phan, Tien M and Kraft, Emma J and Mittal, Jeetain and Showalter, Scott A}, journal = {The Journal of Physical Chemistry B}, volume = {128}, number = {43}, pages = {10636--10649}, year = {2024}, publisher = {ACS Publications}, doi = {https://doi.org/10.1021/acs.jpcb.4c05701}, url = {https://doi.org/10.1021/acs.jpcb.4c05701}, dimensions = {1181580542}, }
2023
- Biophys. J.
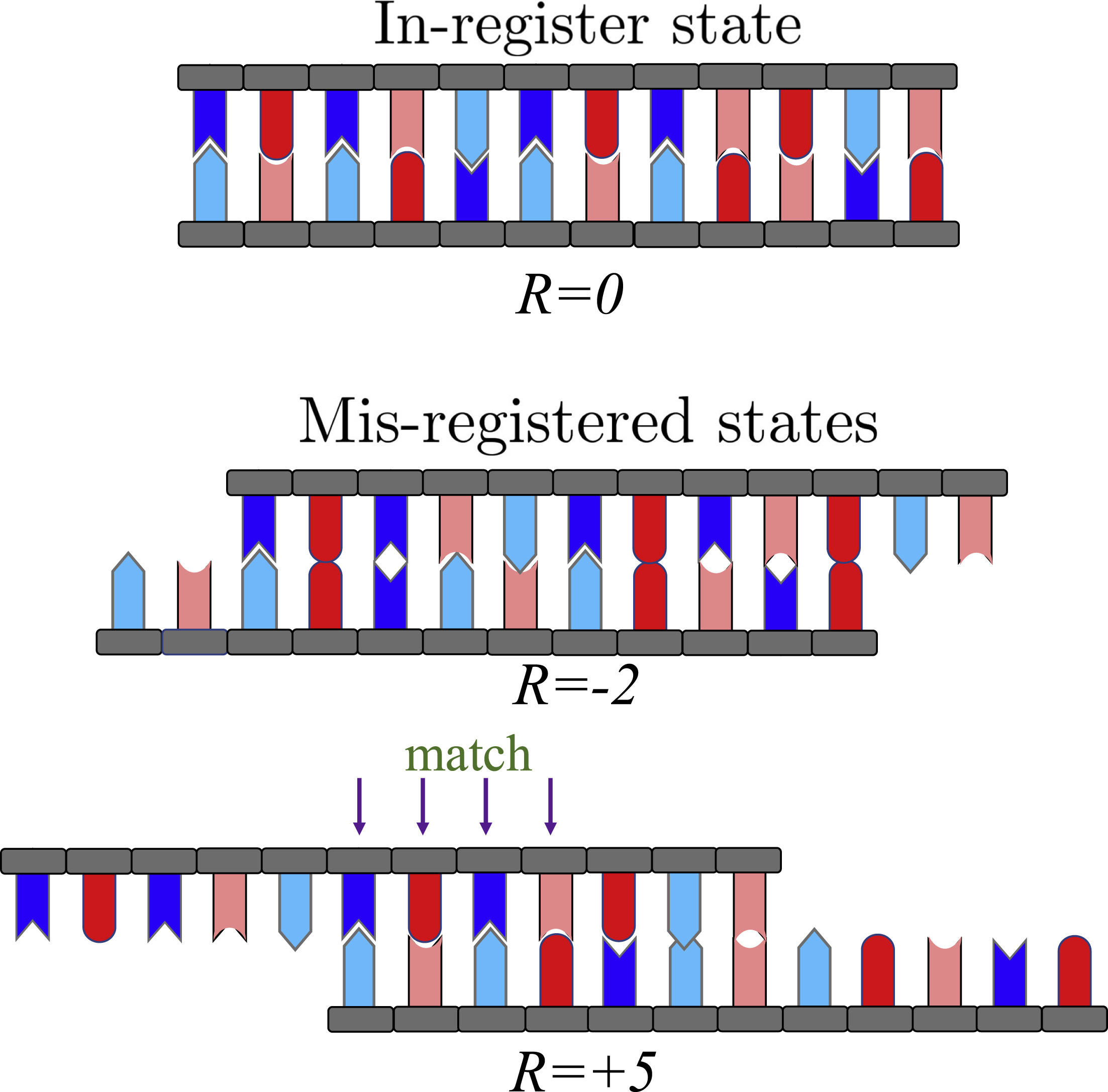 Beneficial and detrimental effects of non-specific binding during DNA hybridizationTam TM Phan, Tien M Phan, and Jeremy D SchmitBiophysical Journal, 2023
Beneficial and detrimental effects of non-specific binding during DNA hybridizationTam TM Phan, Tien M Phan, and Jeremy D SchmitBiophysical Journal, 2023DNA strands have to sample numerous states to find the alignment that maximizes Watson-Crick-Franklin base pairing. This process depends strongly on sequence, which affects the stability of the native duplex as well as the prevalence of non-native inter- and intramolecular helices. We present a theory that describes DNA hybridization as a three-stage process: diffusion, registry search, and zipping. We find that non-specific binding affects each of these stages in different ways. Mis-registered intermolecular binding in the registry search stage helps DNA strands sample different alignments and accelerates the hybridization rate. Non-native intramolecular structure affects all three stages by rendering portions of the molecule inert to intermolecular association, limiting mis-registered alignments to be sampled, and impeding the zipping process. Once in-register base pairs are formed, the stability of the native structure is important to hold the molecules together long enough for non-native contacts to break.
@article{phan2023beneficial, title = {Beneficial and detrimental effects of non-specific binding during DNA hybridization}, author = {Phan, Tam TM and Phan, Tien M and Schmit, Jeremy D}, journal = {Biophysical Journal}, volume = {122}, number = {5}, pages = {835--848}, year = {2023}, publisher = {Elsevier}, doi = {https://doi.org/10.1016/j.bpj.2023.01.034}, url = {https://doi.org/10.1016/j.bpj.2023.01.034}, dimensions = {1154965207}, } - Curr. Opin. Chem. Biol.
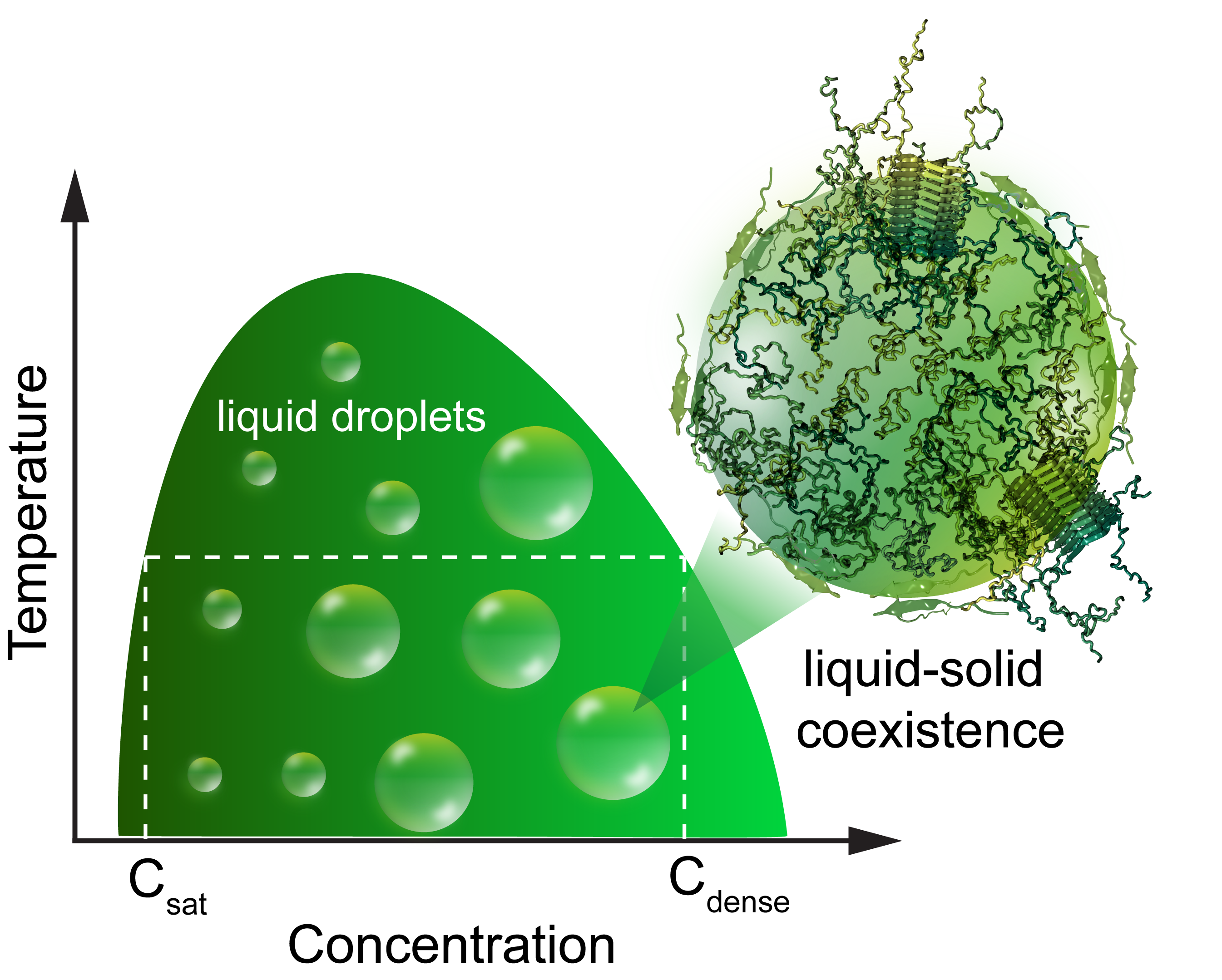 Challenges in studying the liquid-to-solid phase transitions of proteins using computer simulationsBeata Szała-Mendyk, Tien Minh Phan, Priyesh Mohanty, and 1 more authorCurrent opinion in chemical biology, 2023
Challenges in studying the liquid-to-solid phase transitions of proteins using computer simulationsBeata Szała-Mendyk, Tien Minh Phan, Priyesh Mohanty, and 1 more authorCurrent opinion in chemical biology, 2023"Membraneless organelles," also referred to as biomolecular condensates, perform a variety of cellular functions and their dysregulation is implicated in cancer and neurodegeneration. In the last two decades, liquid-liquid phase separation (LLPS) of intrinsically disordered and multidomain proteins has emerged as a plausible mechanism underlying the formation of various biomolecular condensates. Further, the occurrence of liquid-to-solid transitions within liquid-like condensates may give rise to amyloid structures, implying a biophysical link between phase separation and protein aggregation. Despite significant advances, uncovering the microscopic details of liquid-to-solid phase transitions using experiments remains a considerable challenge and presents an exciting opportunity for the development of computational models which provide valuable, complementary insights into the underlying phenomenon. In this review, we first highlight recent biophysical studies which provide new insights into the molecular mechanisms underlying liquid-to-solid (fibril) phase transitions of folded, disordered and multi-domain proteins. Next, we summarize the range of computational models used to study protein aggregation and phase separation. Finally, we discuss recent computational approaches which attempt to capture the underlying physics of liquid-to-solid transitions along with their merits and shortcomings.
@article{szala2023challenges, title = {Challenges in studying the liquid-to-solid phase transitions of proteins using computer simulations}, author = {Sza{\l}a-Mendyk, Beata and Phan, Tien Minh and Mohanty, Priyesh and Mittal, Jeetain}, journal = {Current opinion in chemical biology}, volume = {75}, pages = {102333}, year = {2023}, publisher = {Elsevier}, doi = {https://doi.org/10.1016/j.cbpa.2023.102333}, url = {https://doi.org/10.1016/j.cbpa.2023.102333}, dimensions = {1158506422}, }
2022
- Biochemistry
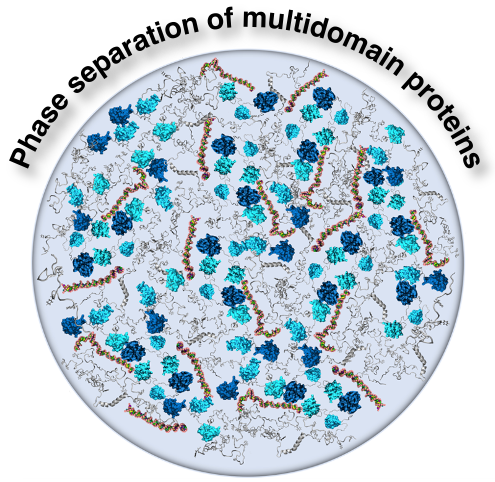 Principles governing the phase separation of multidomain proteinsPriyesh Mohanty, Utkarsh Kapoor, Dinesh Sundaravadivelu Devarajan, and 3 more authorsBiochemistry, 2022
Principles governing the phase separation of multidomain proteinsPriyesh Mohanty, Utkarsh Kapoor, Dinesh Sundaravadivelu Devarajan, and 3 more authorsBiochemistry, 2022A variety of membraneless organelles, often termed “biological condensates”, play an important role in the regulation of cellular processes such as gene transcription, translation, and protein quality control. On the basis of experimental and theoretical investigations, liquid–liquid phase separation (LLPS) has been proposed as a possible mechanism for the origin of biological condensates. LLPS requires multivalent macromolecules that template the formation of long-range, intermolecular interaction networks and results in the formation of condensates with defined composition and material properties. Multivalent interactions driving LLPS exhibit a wide range of modes from highly stereospecific to nonspecific and involve both folded and disordered regions. Multidomain proteins serve as suitable macromolecules for promoting phase separation and achieving disparate functions due to their potential for multivalent interactions and regulation. Here, we aim to highlight the influence of the domain architecture and interdomain interactions on the phase separation of multidomain protein condensates. First, the general principles underlying these interactions are illustrated on the basis of examples of multidomain proteins that are predominantly associated with nucleic acid binding and protein quality control and contain both folded and disordered regions. Next, the examples showcase how LLPS properties of folded and disordered regions can be leveraged to engineer multidomain constructs that form condensates with the desired assembly and functional properties. Finally, we highlight the need for improvements in coarse-grained computational models that can provide molecular-level insights into multidomain protein condensates in conjunction with experimental efforts.
@article{mohanty2022principles, title = {Principles governing the phase separation of multidomain proteins}, author = {Mohanty, Priyesh and Kapoor, Utkarsh and Sundaravadivelu Devarajan, Dinesh and Phan, Tien Minh and Rizuan, Azamat and Mittal, Jeetain}, journal = {Biochemistry}, volume = {61}, number = {22}, pages = {2443--2455}, year = {2022}, publisher = {ACS Publications}, doi = {https://doi.org/10.1021/acs.biochem.2c00210}, url = {https://doi.org/10.1021/acs.biochem.2c00210}, dimensions = {1149335080}, } - Biophys. J.
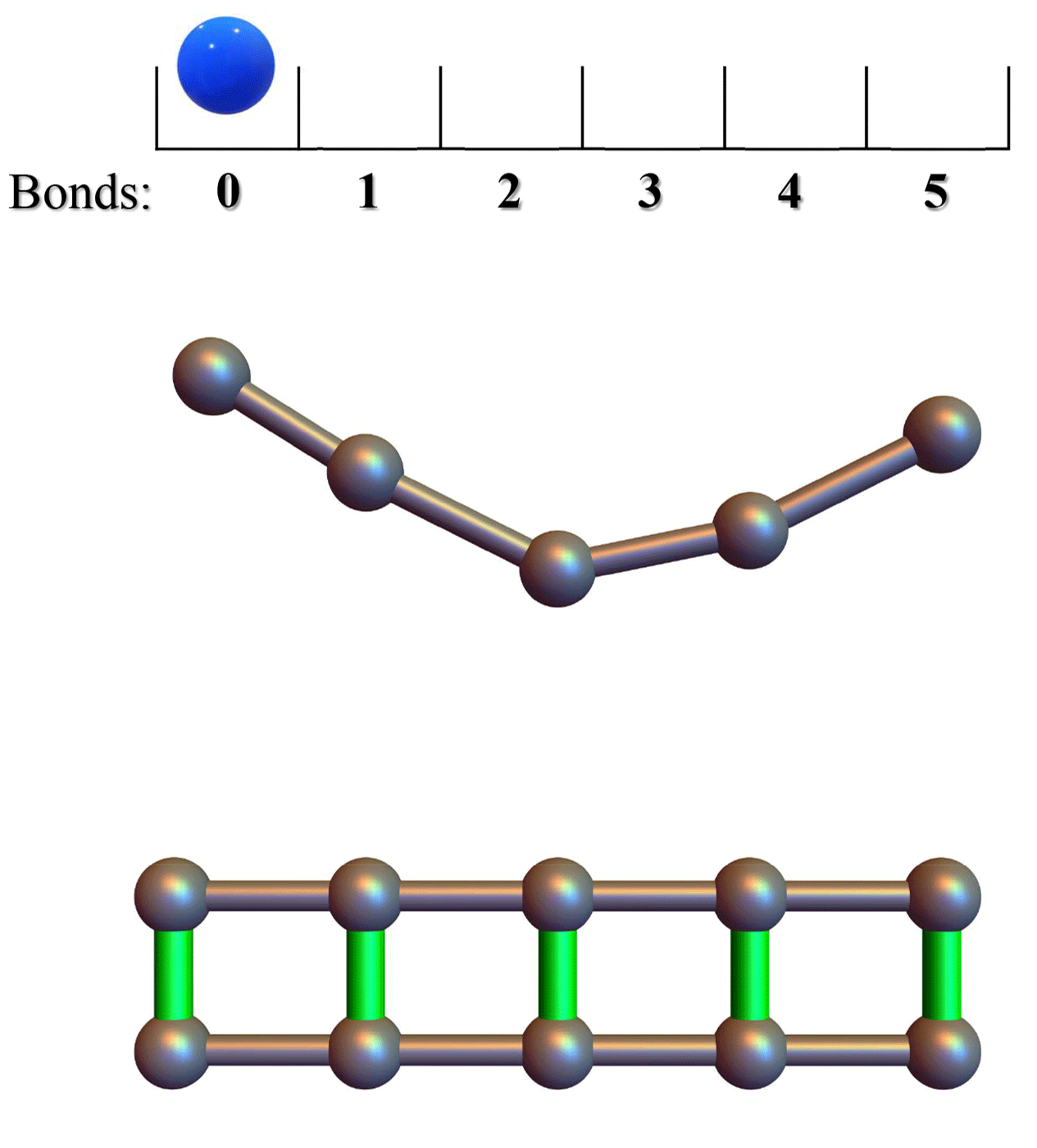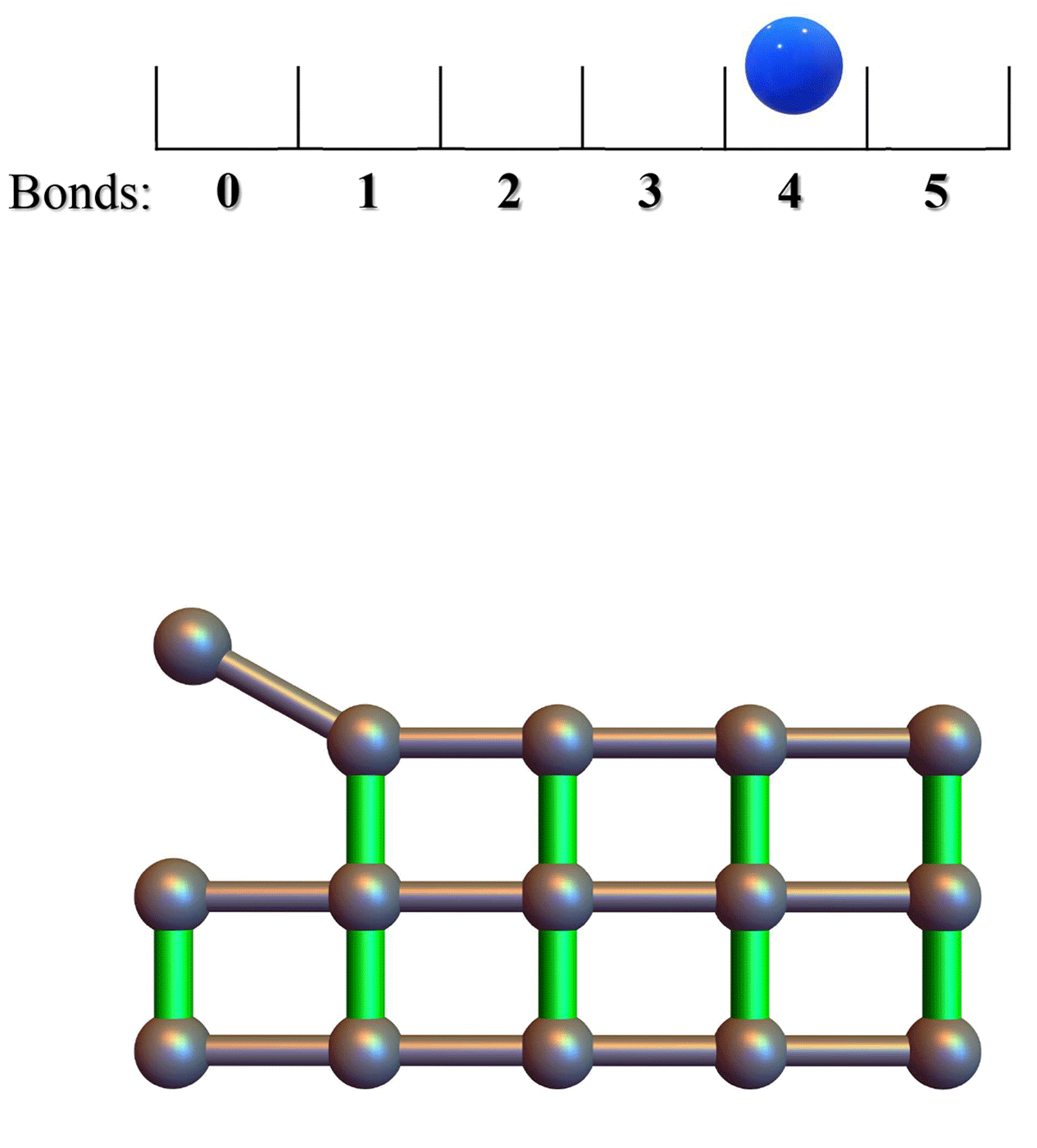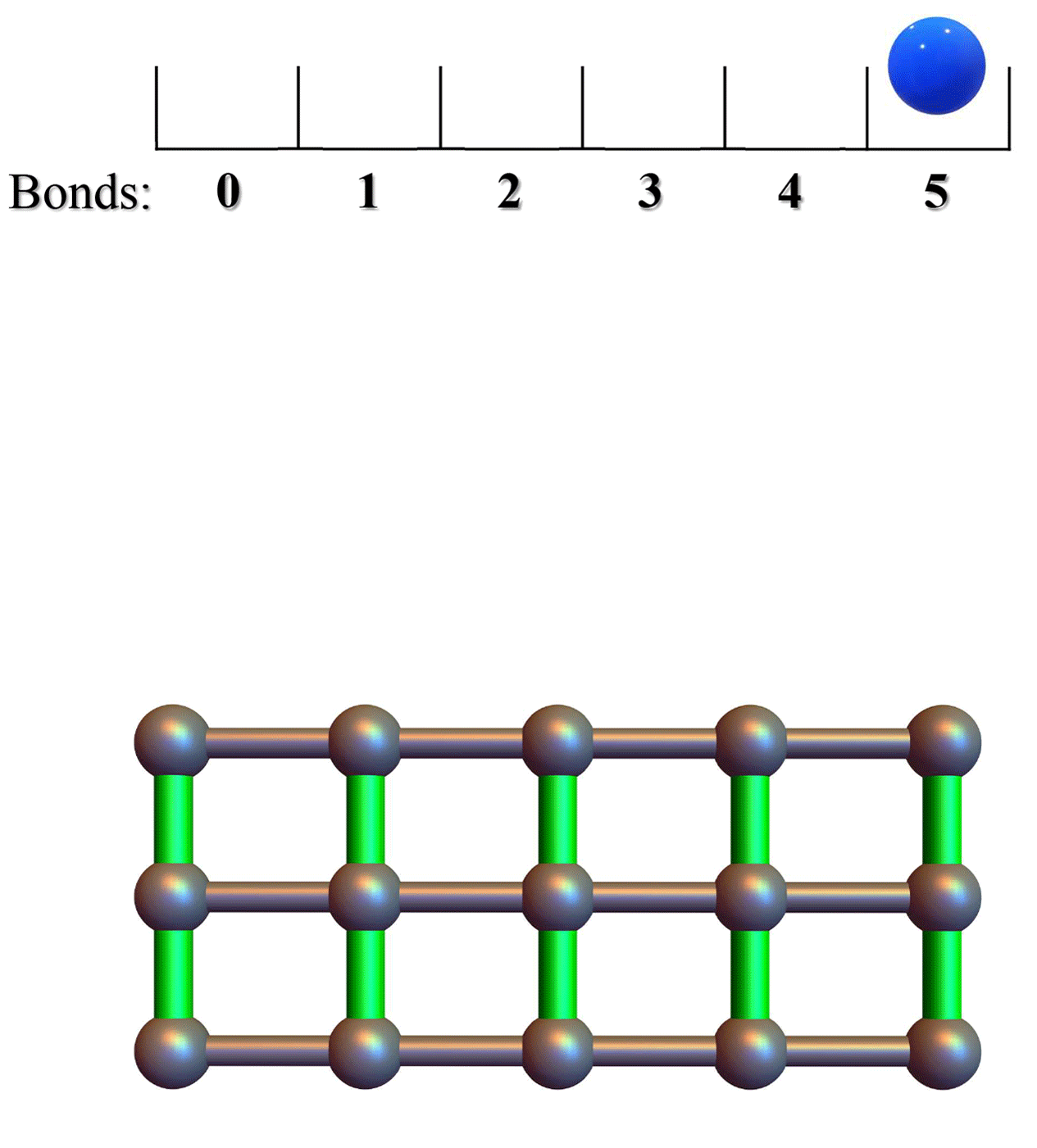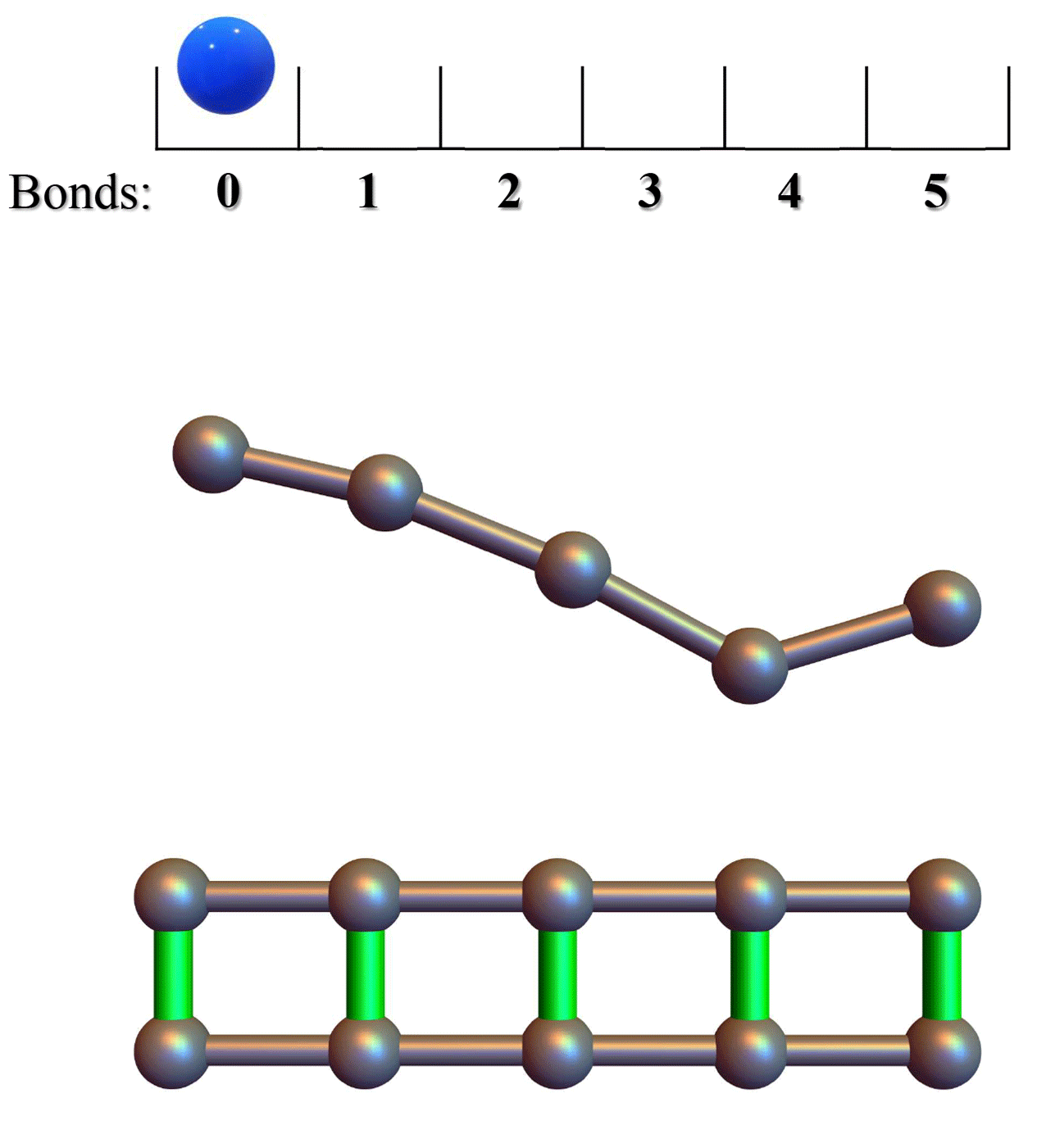 Conformational entropy limits the transition from nucleation to elongation in amyloid aggregationTien M Phan and Jeremy D SchmitBiophysical Journal, 2022
Conformational entropy limits the transition from nucleation to elongation in amyloid aggregationTien M Phan and Jeremy D SchmitBiophysical Journal, 2022The formation of β-sheet-rich amyloid fibrils in Alzheimer’s disease and other neurodegenerative disorders is limited by a slow nucleation event. To understand the initial formation of β-sheets from disordered peptides, we used all-atom simulations to parameterize a lattice model that treats each amino acid as a binary variable with β- and non-β-sheet states. We show that translational and conformational entropy give the nascent β-sheet an anisotropic surface tension that can be used to describe the nucleus with 2D classical nucleation theory. Since translational entropy depends on concentration, the aspect ratio of the critical β-sheet changes with protein concentration. Our model explains the transition from the nucleation phase to elongation as the point where the β-sheet core becomes large enough to overcome the conformational entropy cost to straighten the terminal molecule. At this point the β-strands in the nucleus spontaneously elongate, which results in a larger binding surface to capture new molecules. These results suggest that nucleation is relatively insensitive to sequence differences in coaggregation experiments because the nucleus only involves a small portion of the peptide.
@article{phan2022conformational, title = {Conformational entropy limits the transition from nucleation to elongation in amyloid aggregation}, author = {Phan, Tien M and Schmit, Jeremy D}, journal = {Biophysical Journal}, volume = {121}, number = {15}, pages = {2931--2939}, year = {2022}, publisher = {Elsevier}, doi = {https://doi.org/10.1016/j.bpj.2022.06.031}, url = {https://doi.org/10.1016/j.bpj.2022.06.031}, dimensions = {1149144843}, } - J. Chem. Inf. Model.
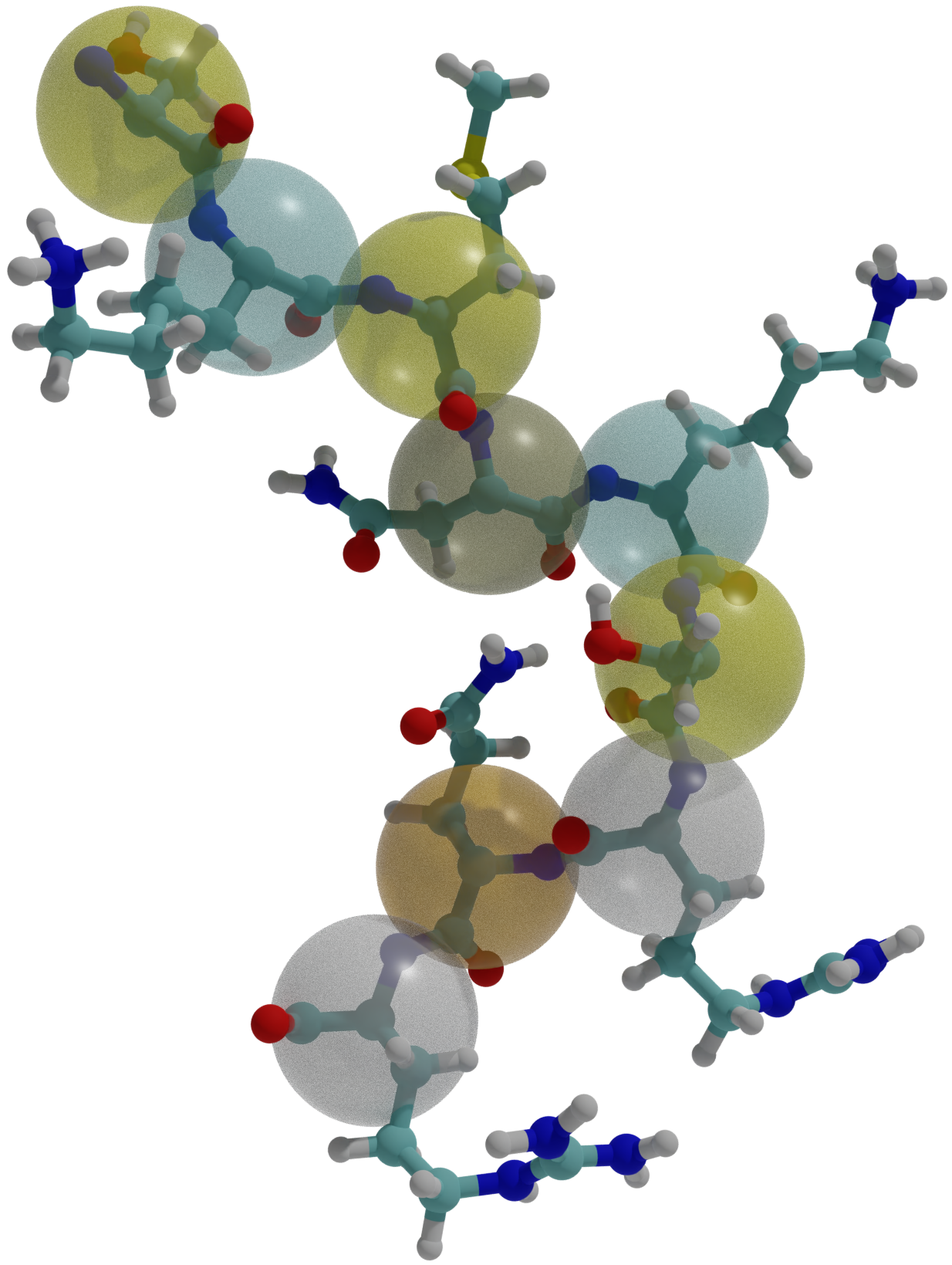 Developing bonded potentials for a coarse-grained model of intrinsically disordered proteinsAzamat Rizuan, Nina Jovic, Tien M Phan, and 2 more authorsJournal of chemical information and modeling, 2022
Developing bonded potentials for a coarse-grained model of intrinsically disordered proteinsAzamat Rizuan, Nina Jovic, Tien M Phan, and 2 more authorsJournal of chemical information and modeling, 2022Recent advances in residue-level coarse-grained (CG) computational models have enabled molecular-level insights into biological condensates of intrinsically disordered proteins (IDPs), shedding light on the sequence determinants of their phase separation. The existing CG models that treat protein chains as flexible molecules connected via harmonic bonds cannot populate common secondary-structure elements. Here, we present a CG dihedral angle potential between four neighboring beads centered at Cα atoms to faithfully capture the transient helical structures of IDPs. In order to parameterize and validate our new model, we propose Cα-based helix assignment rules based on dihedral angles that succeed in reproducing the atomistic helicity results of a polyalanine peptide and folded proteins. We then introduce sequence-dependent dihedral angle potential parameters (εd) and use experimentally available helical propensities of naturally occurring 20 amino acids to find their optimal values. The single-chain helical propensities from the CG simulations for commonly studied prion-like IDPs are in excellent agreement with the NMR-based α-helix fraction, demonstrating that the new HPS-SS model can accurately produce structural features of IDPs. Furthermore, this model can be easily implemented for large-scale assembly simulations due to its simplicity.
@article{rizuan2022developing, title = {Developing bonded potentials for a coarse-grained model of intrinsically disordered proteins}, author = {Rizuan, Azamat and Jovic, Nina and Phan, Tien M and Kim, Young C and Mittal, Jeetain}, journal = {Journal of chemical information and modeling}, volume = {62}, number = {18}, pages = {4474--4485}, year = {2022}, publisher = {ACS Publications}, doi = {https://doi.org/10.1021/acs.jcim.2c00450}, url = {https://doi.org/10.1021/acs.jcim.2c00450}, dimensions = {1150792227}, } - NAR
 Molecular interactions underlying the phase separation of HP1α: role of phosphorylation, ligand and nucleic acid bindingCheenou Her, Tien M Phan, Nina Jovic, and 6 more authorsNucleic Acids Research, 2022
Molecular interactions underlying the phase separation of HP1α: role of phosphorylation, ligand and nucleic acid bindingCheenou Her, Tien M Phan, Nina Jovic, and 6 more authorsNucleic Acids Research, 2022Heterochromatin protein 1α (HP1α) is a crucial element of chromatin organization. It has been proposed that HP1α functions through liquid-liquid phase separation (LLPS), which allows it to compact chromatin into transcriptionally repressed heterochromatin regions. In vitro, HP1α can undergo phase separation upon phosphorylation of its N-terminus extension (NTE) and/or through interactions with DNA and chromatin. Here, we combine computational and experimental approaches to elucidate the molecular interactions that drive these processes. In phosphorylation-driven LLPS, HP1α can exchange intradimer hinge-NTE interactions with interdimer contacts, which also leads to a structural change from a compacted to an extended HP1α dimer conformation. This process can be enhanced by the presence of positively charged HP1α peptide ligands and disrupted by the addition of negatively charged or neutral peptides. In DNA-driven LLPS, both positively and negatively charged peptide ligands can perturb phase separation. Our findings demonstrate the importance of electrostatic interactions in HP1α LLPS where binding partners can modulate the overall charge of the droplets and screen or enhance hinge region interactions through specific and non-specific effects. Our study illuminates the complex molecular framework that can fine-tune the properties of HP1α and that can contribute to heterochromatin regulation and function.
@article{her2022molecular, title = {Molecular interactions underlying the phase separation of HP1$\alpha$: role of phosphorylation, ligand and nucleic acid binding}, author = {Her, Cheenou and Phan, Tien M and Jovic, Nina and Kapoor, Utkarsh and Ackermann, Bryce E and Rizuan, Azamat and Kim, Young C and Mittal, Jeetain and Debelouchina, Galia T}, journal = {Nucleic Acids Research}, volume = {50}, number = {22}, pages = {12702--12722}, year = {2022}, publisher = {Oxford University Press}, doi = {https://doi.org/10.1093/nar/gkac1194}, url = {https://doi.org/10.1093/nar/gkac1194}, dimensions = {1153831641}, }
2019
- PRE
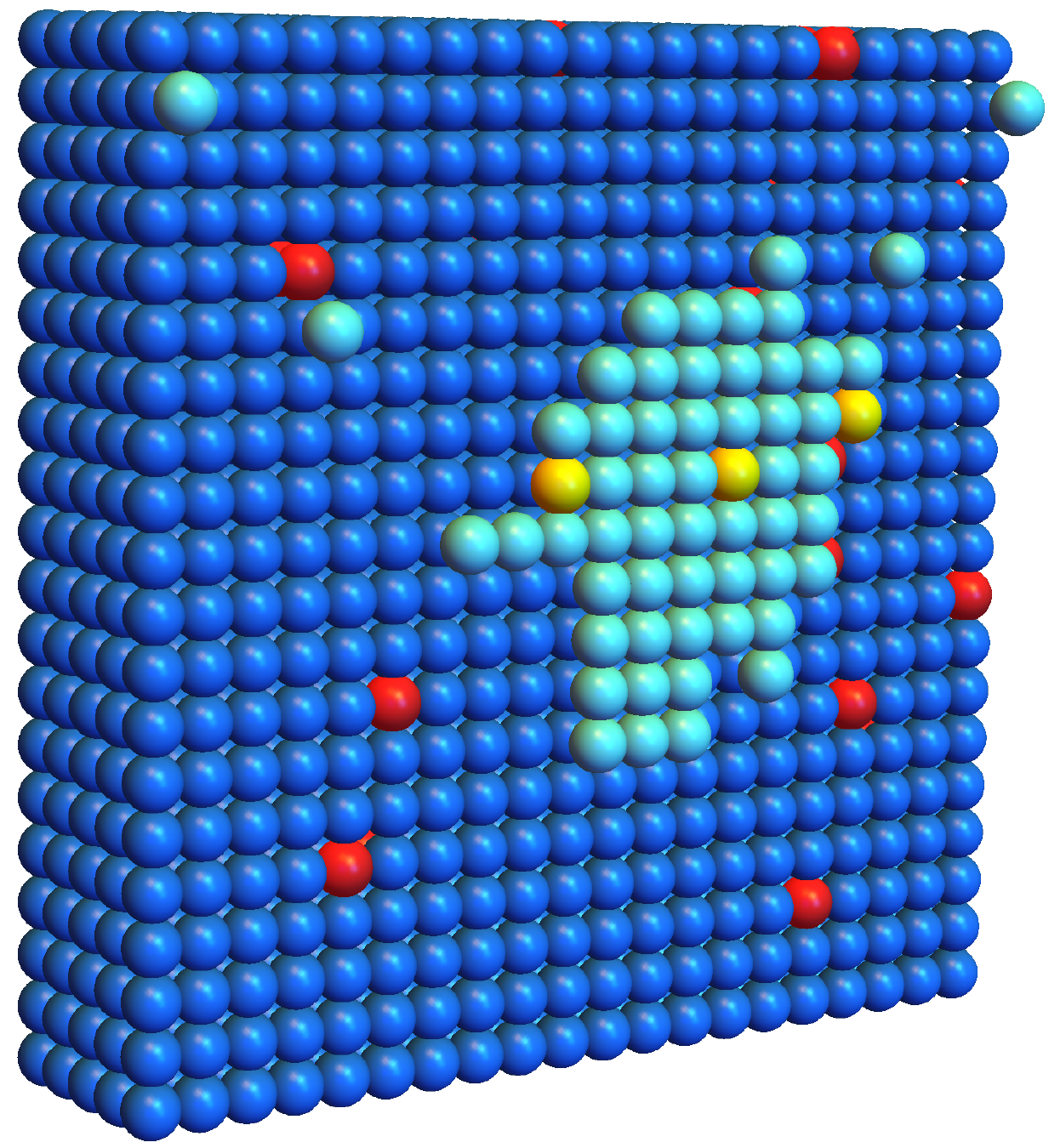 Catalystlike role of impurities in speeding layer-by-layer growthM. Tien Phan, Stephen Whitelam, and D. Jeremy SchmitPhysical Review E, 2019
Catalystlike role of impurities in speeding layer-by-layer growthM. Tien Phan, Stephen Whitelam, and D. Jeremy SchmitPhysical Review E, 2019Molecular self-assembly is usually done at low supersaturation, leading to low rates of growth, in order to allow time for binding mistakes to anneal. However, such conditions can lead to prohibitively long assembly times where growth proceeds by the slow nucleation of successive layers. Here we use a lattice model of molecular self-assembly to show that growth in this regime can be sped up by impurities, which lower the free-energy cost of layer nucleation. Under certain conditions impurities behave almost as a catalyst in that they are present at high concentration at the surface of the assembling structure, but at low concentration in the bulk of the assembled structure. Extrapolation of our numerics using simple analytic arguments suggests that this mechanism can reduce growth times by orders of magnitude in parameter regimes applicable to molecular systems.
@article{phan2019catalystlike, title = {Catalystlike role of impurities in speeding layer-by-layer growth}, author = {Phan, M. Tien and Whitelam, Stephen and Schmit, D. Jeremy}, journal = {Physical Review E}, volume = {100}, number = {4}, pages = {042114}, year = {2019}, publisher = {APS}, doi = {https://doi.org/10.1103/PhysRevE.100.042114}, url = {https://link.aps.org/doi/10.1103/PhysRevE.100.042114}, dimensions = {1121790674}, }